CRISPR genome editing
Overview
CRISPR genome editing is a technology that enables precise, intentional, and permanent changes in the DNA of living organisms. This cost-effective, rapid, and easy-to-use technology relies on Cas enzymes and guide RNAs that are derived from a naturally occurring mechanism in bacterial immune systems.

CRISPR: A POWERFUL TOOL FOR EDITING GENOMES
CRISPR stands for clustered regularly interspaced short palindromic repeats. The term “repeats” refers to repetitive DNA sequences found in bacterial genomes. These unusual DNA sequences were first found within the genome of E. coli in 1987 and are now known to be part of a bacterial adaptive defense mechanism against invading bacterial viruses (bacteriophages) [1]. These repeats are short sequences separating viral DNA sequences that are integrated into the bacterial genome after a previous infection. This bacterial immune system also involves Cas (CRISPR-associated) enzymes—naturally occurring bacterial enzymes that cut the DNA of invading viruses. The CRISPR sequences recognize the DNA of a newly invading virus, facilitating its destruction by the Cas enzyme.
In 2013, researchers demonstrated that CRISPR-Cas–dependent genome editing can be performed in mammalian cells, igniting the CRISPR revolution [2,3].
To apply CRISPR technology in the lab, researchers design a small RNA molecule, including a short sequence that matches a target sequence within a genome. This RNA molecule, known as guide RNA (gRNA), binds to the Cas enzyme and guides it to the target sequence where editing will take place. For CRISPR genome editing, scientists have introduced Cas enzymes in mammalian, plant, fish, worm, as well as other cell types or organisms.
HOW TO USE CRISPR
CRISPR technology gives scientists the ability to add, delete, or alter the genetic material at a location within the genome of a target cell or organism. The technology has the potential to provide benefits for disease research and improve crops and animals to help feed the growing world population.
The most common applications include:
- Knocking out genes
- Introducing or correcting mutations
- Modifying gene expression
- Inserting, modifying, or labeling DNA
- Screening for genes of interest
CRISPR genome editing has been applied primarily to basic biomedical research. Genome editing is also being applied in areas such as agriculture for improving crop yields and increasing disease resistance of crops or livestock, and in industrial biotechnology for improving biofuels and producing other biomaterials used in multiple industries.
The CRISPR basics handbook
Everything you need to know about CRISPR, from A to Z, from theory to practice, for beginners as well as advanced users.
HOW CRISPR WORKS
CRISPR technology relies on two components, a Cas nuclease and a guide RNA (gRNA), that are delivered into a cell to cut the cell’s genome at a target location. The gRNA is designed to match a target site in the genome that is next to a site called a protospacer-adjacent motif (see below) and guides the Cas enzyme where to precisely cut the genomic DNA. Then cellular DNA repair mechanisms are activated, allowing genomic DNA sequence changes to occur, as described below.
What are Cas enzymes, and what do they do?
Cas enzymes are nucleases, enzymes which cut DNA or RNA. Following the discovery of the CRISPR-Cas system in prokaryotes, it was shown that S. pyogenes Cas9 nuclease could be directed by a gRNA (see description below) to cleave genomic DNA at target locations. Many Cas enzymes have now been isolated from a variety of bacterial species. Cas9 and Cas12a (formerly known as Cpf1), which both cut DNA, are well characterized and commonly used nucleases in genome editing applications. The Cas13 family has been shown to target RNA.
What is a guide RNA, and how does it work?
Guide RNA (gRNA) refers to RNA molecules that guide the Cas enzyme to a set target site within the genome. Depending on the Cas enzyme used, gRNA could comprise one or two RNA molecules.
The most commonly used Cas enzyme, Cas9, naturally employs two RNA molecules together: the CRISPR RNA (crRNA) and the transactivating CRISPR RNA (tracrRNA). A section of the tracrRNA forms a duplex with a section of the crRNA (Figure 1A). The crRNA molecule specifies the target DNA by base-pairing with the target sequence. The tracrRNA molecule activates the Cas9 endonuclease enzyme. However, for laboratory use, the crRNA and the tracrRNA can be fused via a hairpin loop into one longer RNA molecule known as single guide RNA (sgRNA) (Figure 1B).
The other commonly used Cas enzyme, Cas12a, employs only a CRISPR RNA (crRNA), which functions as the gRNA to direct Cas12a to the target sequence (Figure 1C). Cas12a does not use a tracrRNA.
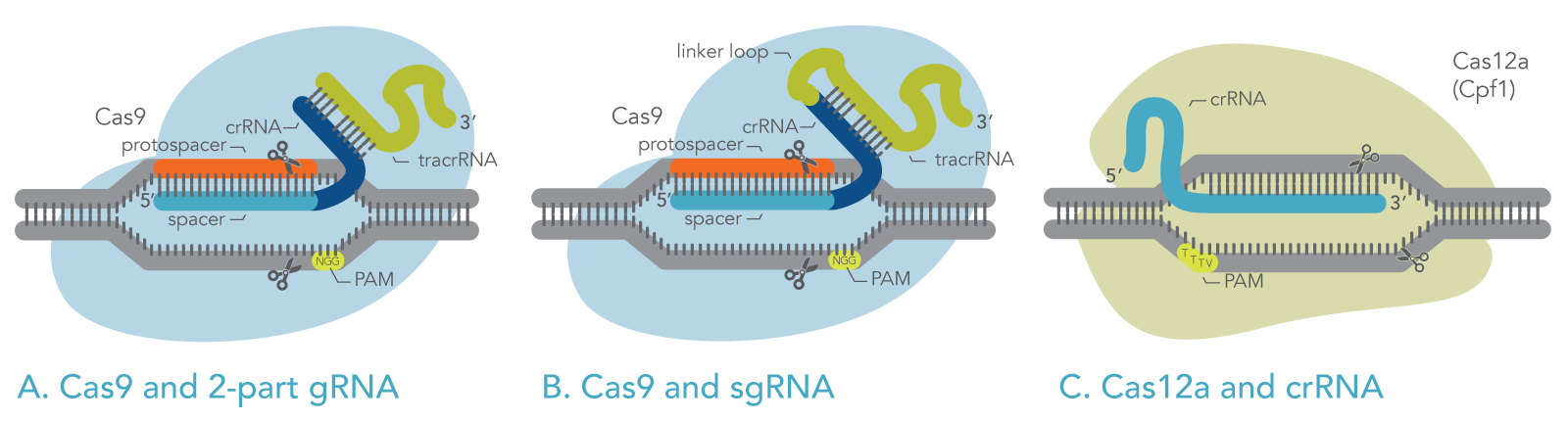
Figure 1. Schematic of active ribonucleoproteins containing Cas enzymes bound to guide RNAs. (A) Cas9 with 2-part guide RNA, comprising crRNA and tracrRNA. (B) Cas9 with single-guide RNA. (C) Cas12a with crRNA (gRNA).
The gRNA, for either Cas9 or Cas12a, contains a target-specific sequence (the “spacer”). The spacer comprises 20 bases for Cas9, or 21 bases for Cas12a. This sequence is complementary to the target DNA. The gRNA spacer pairs with the target DNA sequence, the “protospacer,” which is the section of the genomic DNA that will be cut. For Cas9 and Cas12a to function, the protospacer (target) must be located next to a protospacer adjacent motif (PAM) sequence, as described below.
What is a PAM sequence, and what does it do?
To bind and cleave the target DNA, Cas enzymes require that the target be immediately adjacent to a short PAM sequence, which varies depending on the Cas enzyme. For example, Cas9 derived from S. pyogenes recognizes the PAM sequence 5ʹ-NGG-3ʹ (see Figure 1) [4,5], and Cas12a from Acidaminococcus sp. recognizes the PAM sequence 5ʹ-TTTV-3ʹ (where V represents A, G, or C), as shown in Figure 1.
How are the Cas enzyme and the gRNA produced for experimental use?
Some researchers have produced these components by expression from plasmids transfected into the experimental cells. Others have produced gRNA by in vitro transcription (IVT). However, more recent research has shown significantly better outcomes in terms of the ratio of on/off-target editing when the CRISPR components are prepared in advance and delivered as intact ribonucleoprotein (RNP). Cas enzymes can be produced by standard recombinant protein technology and the gRNA by chemical synthesis. To form the RNP, the Cas and the gRNA are combined outside the cells. The RNP can then be introduced into cells by electroporation or a few other methods. Transfection efficiency by electroporation can be increased by adding electroporation enhancers, which are thought to act as carrier DNA.
CRISPR WORKFLOW:
- Design—design gRNA (and HDR donor template, if needed) for the Cas enzyme chosen for your cell type and conditions.
- Deliver—introduce CRISPR reagents into cells.
- Repair—allow cells to undergo editing.
- Analyze—determine (mismatch cleavage assay) or quantify (rhAmpSeq CRISPR Analysis System) editing results.
Products for CRISPR genome editing
Alt-R™ CRISPR-Cas9 System
Efficient CRISPR reagents based on the commonly used Streptococcus pyogenes Cas9 system for lipofection or electroporation experiments. Protospacer adjacent motif (PAM) = NGG.
Alt-R™ CRISPR-Cas12a (Cpf1) System
For additional target sites or for targeting AT-rich regions, use the CRISPR-Cas12a system in electroporation experiments. Protospacer adjacent motif (PAM) = TTTV. The Alt-R Cas12a (Cpf1) Ultra also can recognize many TTTT PAM sites in addition to TTTV motifs, increasing target range for genome editing studies.
Alt-R™ CRISPR nucleases
Recombinant, high-purity Cas9 and Cas12a (Cpf1) endonucleases for genome editing experiments.
Alt-R™ HDR Donor Oligos
Single-stranded DNA oligos specifically built for your homology‑directed repair (HDR) experiments. Designed and optimized through extensive laboratory testing, they are ideal for introducing point mutations and short insertions.
Alt-R™ HDR Donor Blocks
Double-stranded DNA fragments specifically built for your homology-directed repair (HDR) experiments. IDT dsDNA templates offer a cost-effective, high-fidelity option to reduce the amount of downstream screening via high HDR rates and low unintended blunt integrations. Alt-R HDR Donor Blocks are ideal for making large insertions or genomic changes.
rhAmpSeq™ CRISPR Analysis System
An end-to-end solution to design, deploy, and analyze next generation sequencing data for on- and off-target interrogation after your CRISPR experiment.
Alt-R™ Genome Editing Detection Kit
T7 endonuclease I (T7EI) mismatch cleavage assay for detection of on-target editing, known off-target events, and estimation of genome editing efficiency in cultured cells.
Explore resources
DECODED articles
References
- Lander ES. The Heroes of CRISPR. Cell. 2016;164(1-2):18-28.
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819-823.
- Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823-826.
- Anders C, Niewoehner O, Duerst A, et al. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513(7519):569-573.
- Sternberg SH, Redding S, Jinek M, et al. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507(7490):62-67.
RUO22-1441_001