
The genetic mutation that causes the sickle hemoglobin
Sickle cell disease (SCD) is the most common genetic disease in the US and affects approximately 100,000 Americans and more than 3 million people worldwide [2]. Each year, there are 300,000 babies born with SCD [3]. SCD is caused by point mutations occurring in the beta hemoglobin gene. These mutations can cause a single amino acid substitution in the beta globin protein recognized as sickle hemoglobin or hemoglobin S (HbS) and leading to polymerization and sickling of red blood cells (RBCs). Hemoglobin helps RBCs facilitate oxygen delivery from the lungs to various tissues and organs. Sickle hemoglobin delivers less oxygen between the lungs and tissues. Sickle cells adhering to the endothelium and persistent inflammation creates a vicious cycle leading to vasoocclusive crisis (VOC) in SCD [4]. SCD patients experience recurrent pain crises which significantly impacts their quality of life.
The role of fetal hemoglobin in sickle cell disease
The fetal-to-adult hemoglobin switch occurs after birth. In adults, fetal hemoglobin (HbF) levels are less than 1% of total hemoglobin [5]. There are two distinct globin chains each with its own heme molecule which combines to form hemoglobin. In the fetus, there are distinct non-alpha chains called gamma. The two gamma chains and the two alpha chains form what is known as fetal hemoglobin (HbF) which is the primary hemoglobin expressed in the developing fetus.
Within 18 to 24 weeks post birth, these non-alpha globin chains are replaced with what is referred to as beta globin. The two alpha chains and two beta chains are what is known as adult hemoglobin (HbA). The pairing of one non-alpha and one alpha to form a hemoglobin dimer does not efficiently deliver oxygen. Two dimers form the tetramer which is essential for oxygen transport from the lungs to the tissues and organ systems in the body.
In SCD, an elevated presence of HbF reduces the sickling [6]. In fact, newborns with SCD have fewer sickle cells due to a higher concentration of fetal hemoglobin in their circulatory system.
BCL11A functions as the molecular switch which has a direct role in transitioning fetal to adult hemoglobin
Genome wide association studies (GWAS) have revealed that single nucleotide polymorphisms (SNPs) in the B-cell lymphoma/leukemia 11A (BCL11A) gene on chromosome 2 contribute to variations of fetal hemoglobin expression [7]. BCL11A gene is a transcription factor that has been shown to repress fetal hemoglobin expression. The function of BCL11A serves as the molecular switch for transitioning fetal hemoglobin (HbF) to beta hemoglobin also referred to as adult hemoglobin (HbA). Thus, knocking out BCL11A increases HbF and represses the sickle hemoglobin [8].
How CRISPR-Cas9 gene editing is performed to target BCL11A to induce fetal hemoglobin in sickle cell disease
Vertex Pharmaceuticals and CRISPR Therapeutics® developed and received approvals for CRISPR CTX001™ gene editing therapy both in the EU and the US to treat SCD. CTX001 is an autologous, ex vivo CRISPR- Cas9 gene editing therapy which successfully induces HbF in the RBCs using the patient’s hematopoietic stem cells (HPSCs) [9]. The discovery of an enhancer region of BCL11A specific to the erythroid linage allowed researchers to design guide RNAs with a target sequence for BCL11A [10]. Repressing BCL11A, promotes the expression of fetal hemoglobin which restores adult hemoglobin (Figure 1).
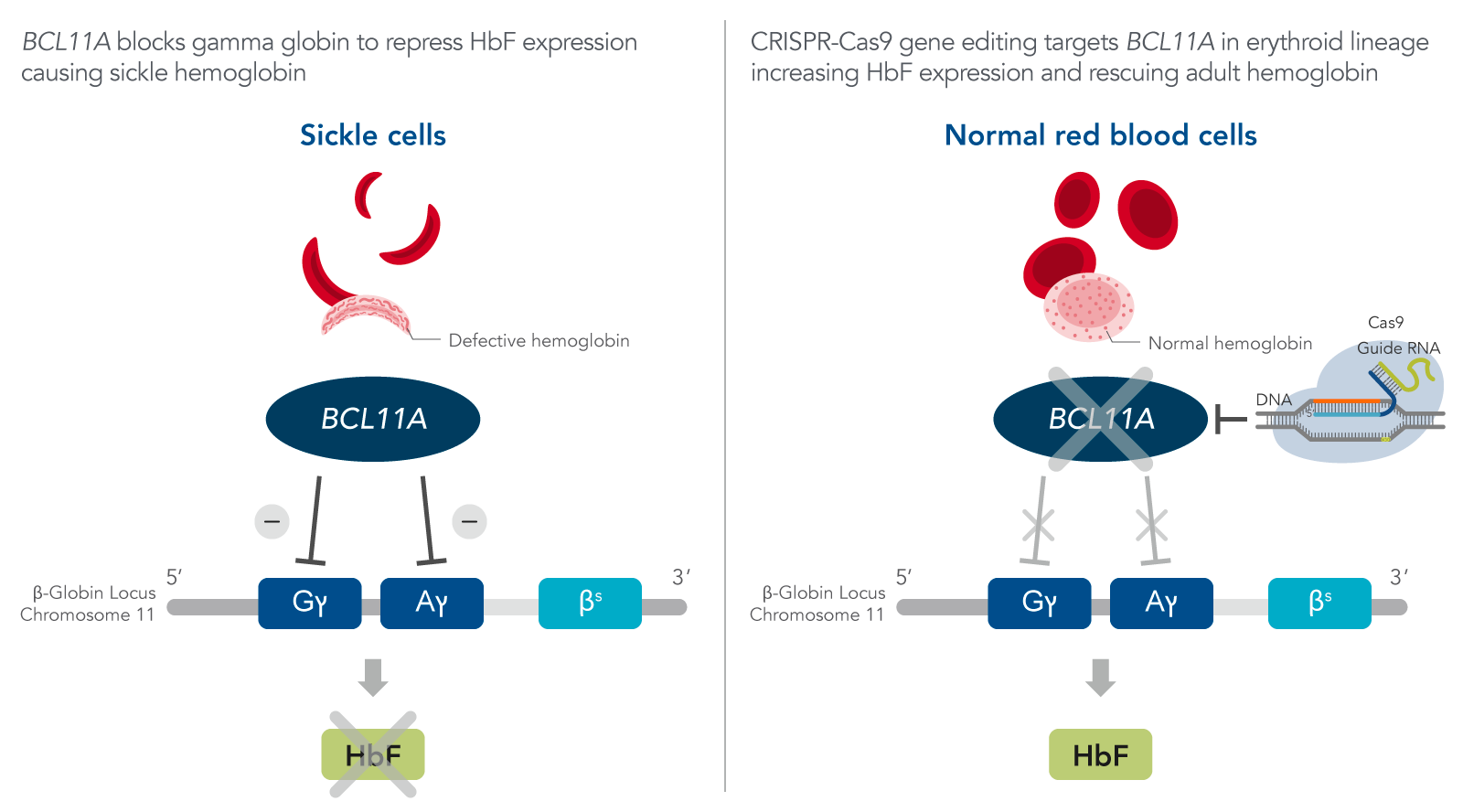
Figure 1. FDA-approved CRISPR-Cas9 gene therapy for sickle cell disease. The beta globin locus is arranged sequentially from 5’ to 3’ starting with gamma and ends with the adult beta globin. There are two copies of the gamma genes on each chromosome 11, while beta is presented as a single copy on the chromosome. BCL11A represses fetal hemoglobin (HbF) expression. Therefore, the guide RNA designed specifically targets BCL11A resulting its disruption in the erythroid lineage which leads to increase HbF expression and rescues the adult beta hemoglobin.
Future outlook for treating SCD using CRISPR-Cas9 therapy
Using CRISPR-Cas9 gene therapy, SCD patients can now live a better quality of life without being affected by their genetic predisposition. Studies are underway to correct the SCD causing mutation. The Phase I SCD clinical trial lead by Dr. Matthew Porteus from Standford Medicine is evaluating a gene editing therapy to fully restore the disease-causing sickle HBB allele using a high fidelity Cas9 enzyme [11]. With the highly successful application of CRISPR therapy, researchers and clinicians can apply this technology to treat various genetic diseases.
In efforts to provide highly efficient CRISPR therapeutic products, IDT recently announced the opening of our dedicated Therapeutic Oligonucleotide Manufacturing Facility to produce cGMP guide RNA. IDT also partnered with Aldevron to offer a single vendor solution for high-quality consistent CRISPR genome editing tools to help scientists in their journey from discovery to clinic.
Download our CRISPR Therapeutics eBook
CRISPR Therapeutics (PDF) is designed to explore the vast possibilities of Genomic Medicine. It will help translational research and cell and gene therapy developers discover what we have to offer with our cGMP/q7 capabilities for developing CRISPR-Cas9 gene therapies.
Download your copy of CRISPR Therapeutics eBook.

