
Cloning of double-stranded DNA (dsDNA) molecules into plasmid vectors is a commonly employed molecular biology technique. The procedure is used for sequencing, building libraries of DNA molecules, expressing coding and non-coding RNA, and many other applications. The purpose of this article is to discuss cohesive end cloning—one method by which DNA fragments can be inserted into a plasmid vector using restriction digestion.
Creating cohesive ends
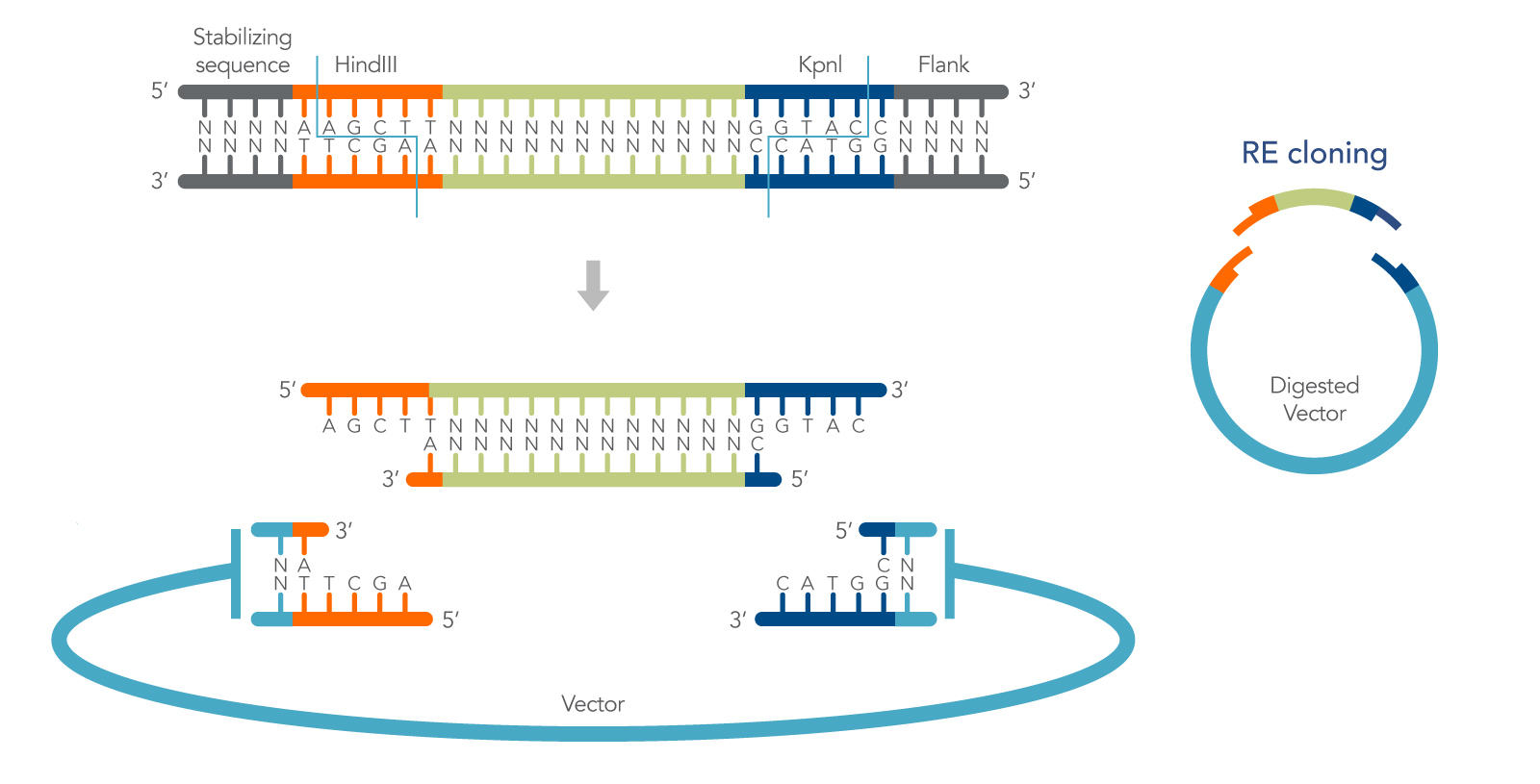
Restriction endonucleases recognize and cleave dsDNA at highly specific nucleotide sequences, or restriction sites. Many restriction enzymes produce short, staggered ends (with 5′ or 3′ overhangs) that can be rejoined by a DNA ligase to regenerate the original recognition sequence (Figure 1). They are referred to as cohesive ends because of the hydrogen bond stabilization of the DNA bases that loosely holds the DNA ends together prior to ligation. The specificity of restriction enzymes enables directional cloning, and the hydrogen bonding of cohesive ends increases the efficiency of cohesive end ligation by as much as 100X over blunt-end ligation [1]. These features make cohesive end cloning a highly useful method for molecular biology.
Cohesive end restriction cloning can be described in a relatively standard series of steps:
- First, the insert is designed with restriction sites that also occur in the vector multiple cloning site (MCS), but not elsewhere in the insert or vector.
- Second, the insert and plasmid are digested in separate reactions, using the chosen enzyme(s). Following digestion, the plasmid is dephosphorylated, and both insert, and plasmid are purified to remove all remaining enzymes.
- In the remaining steps the insert is ligated into the plasmid, and the ligated plasmid is transformed into E. coli. Below, the method is described in more detail with added tips that further increase its flexibility.
Plasmids 101: What are plasmids?
Plasmids are one of the most common types of vectors used for amplifying and introducing DNA into cells. Plasmids are double-stranded, circular, extrachromosomal DNA molecules that contain the necessary DNA elements required for replication in E. coli cells.
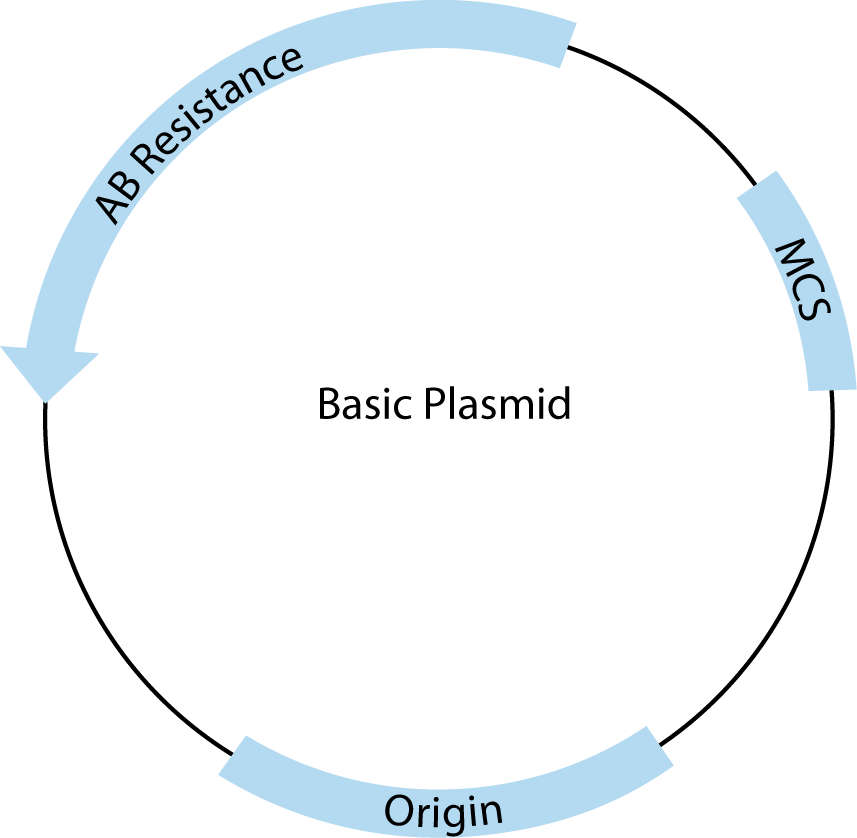
All plasmids must contain three basic features to be useful for cloning. A schematic drawing of a basic cloning vector is shown in Figure 2.
The first required feature is the origin of replication, which recruits endogenous bacterial replication machinery to copy transformed plasmids. The number of copies of a plasmid produced within a single E. coli is a direct result of the type of origin used. For example, pUC contains a high copy origin, producing 500–700 copies per cell under ideal conditions [1]. The endogenous E. coli replication machinery produces these copies with very high fidelity.
The second is the multiple cloning site (MCS), which contains closely spaced restriction enzyme recognition sequences that occur only in the MCS and can be used to insert one or more pieces of DNA in this region without disrupting the rest of the plasmid structure.
Third, plasmids must contain at least one antibiotic resistance gene (AB Resistance). This enables the small percentage of bacteria that have taken up and copied the transfected plasmid to be selected using agar plates containing the specified antibiotic. For example, the β-lactamase gene, included in many plasmids, confers resistance to ampicillin. E.coli that contains a complete, circular plasmid with this gene will make small, isolated, circular colonies on ampicillin-containing, agar plates that can be easily picked and used to produce large amounts of that specific, cloned plasmid.
In addition to these basic features, many commercial plasmids contain one or more features that facilitate cloning, regulate expression of experimental inserts, or add features to expressed RNA or proteins, making them one of the most versatile tools in molecular biology. These added elements are almost always adjacent to the MCS. For most common applications, there is more than one appropriate vector choice, so it is important to understand the features you need and to compare those with the advantages and disadvantages of the available vector tools.
Plasmid inserts
The plasmid vectors used for general cloning are usually 3–6 kb in length. The sizes of inserts they can accommodate and replicate are typically <5 kb, although specialized plasmids may allow for inserts up to 10 kb or more. Check with the manufacturer of the plasmid for the recommended insert size range. In general, smaller inserts are easier to clone than larger ones. Longer sequences will also naturally contain more endogenous restriction sites than shorter inserts, limiting the enzymes that can be used for cloning without destroying the insert.
Several free online software programs are available to identify restriction sites within the plasmid and insert sequences, including Webcutter, RestrictionMapper, and NEBcutter. In some cases the MCS itself may need to be altered—e.g., to add or remove certain restriction sites before the experimental DNA can be cloned. This can be done by site-directed mutagenesis, blunt-end cloning, or seamless cloning to avoid this issue (see The mutagenesis handbook).
Considerations for cohesive end experiments
In addition to providing efficient ligations, and the ability to clone directionally using double digests, another outcome of cohesive end cloning is that ligation of the insert into the vector recreates the original restriction sites. This can be useful for screening clones or for the addition of adjacent DNA sequences in a subsequent round of cloning.
It is also important to note that most restriction enzymes will not cut a recognition sequence that is at, or very near, the end of the DNA. Therefore, when selecting or engineering your restriction sites, it is important to include a few nonspecific bases, sometimes referred to as buffers, at the end of your insert. These allow the enzyme to bind the DNA efficiently prior to cleavage. Buffer length varies by enzyme and should be confirmed as part of the experimental design process. Also, avoid using restriction sites that are right next to each other in the MCS—a 12 bp buffer between cut sites is generally sufficient for most enzymes [1].
Reaction conditions
The activity of enzymes is typically defined in units, with one unit being the amount of enzyme required to digest 1 µg DNA in 1 hour under the optimal enzyme conditions. Check the manufacturer’s guidelines for how much enzyme to use and the ideal buffer conditions.
For the ligation to be effective, the restriction enzyme digestion must be stopped. Some enzymes are easily heat inactivated by incubation at high temperature for a short period of time. Others cannot be heat inactivated and digestion products have to be purified by column, gel, or phenol extraction methods prior to ligation. Common restriction enzymes that cannot be heat inactivated include BamHI, BclI, and HpaI. Failure to inactivate restriction enzymes will create competing reactions, and produce little or no ligated DNA.
Restriction enzyme reaction conditions are also very important. Salt conditions that are too high or the presence of inhibitors can change the specificity of restriction enzymes. In some cases, this can result in nonspecific cutting, known as star activity, which will generate multiple fragments (detected as multiple unexpected bands on a gel). A common cause of star activity is evaporation of reaction buffer during long digestions, especially those conducted overnight. Perform overnight digestions in sufficient volumes and in small tubes that limit the surface area for evaporation or in a thermal cycler with a heated bonnet to prevent star activity.
Phosphatase and gel purification
If you are digesting with a single enzyme, or one that produces blunt ends, it is important to dephosphorylate the vector using an enzyme such as calf-intestinal alkaline phosphatase (CIAP) or shrimp alkaline phosphatase (SAP). This prevents the vector from religating without incorporating the insert, which still has the requisite phosphates intact. If the ends of the vector can simply religate, the probability of two ends of the same molecule coming together is much higher than the correct ends of a separate vector and insert. Do not dephosphorylate the insert.
Gel purification of the vector removes any uncut vector that will generate background colonies on agar plates. It also removes restriction enzymes and the small DNA fragments cut from MCS that could religate if the vector is not dephosphorylated.
Versatile T4 ligase
The most common method of covalently joining the insert and plasmid DNA ends is to use the ATP-dependent, T4 DNA ligase. Ligation using this enzyme is usually performed between 4–25°C. Cohesive ends have an annealing temperature below room temperature because they are only a few bases long. Low temperature ligations help stabilize DNA interactions, but at the expense of enzyme activity. The reduced activity of T4 ligase at lower temperatures can be partially compensated for by increasing the duration of the ligation reaction.
In addition, because annealing temperature of the ends affects ligation efficiency, cohesive end ligations with short or single base overhangs may benefit from higher concentrations of T4 ligase. Follow the manufacturer’s reaction protocol for their T4 ligase and buffer. For extended incubations, some researchers even add additional enzyme into the reaction midway through.
To generate circular plasmids with the desired insert, and to limit the formation of undesired secondary products like concatemers, the total concentration of DNA in the ligation reaction should be less than 10 µg/mL [1]. Ligations are regularly carried out in the presence of excess insert. For cohesive end cloning, a relatively low insert: vector ratio of 3:1—5:1 should be effective but may need to be experimentally optimized. Figure 3 shows a simple formula for calculating the amount of insert needed.
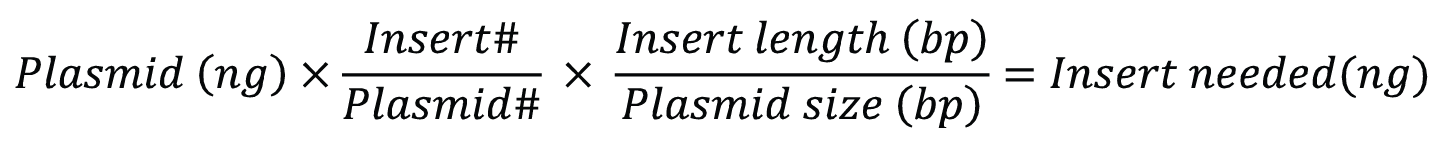
# is the molar ratio of insert to plasmid.
Inhibitors and other problems
Organic solvents including ethanol and phenol are common laboratory reagents that can inhibit restriction enzymes. In addition, the glycerol that most enzymes are shipped in can inhibit reactions if the concentration is too high (above 5%).
Bacteria also naturally inhibit cleavage at certain sites with endogenous Dam and Dcm methyltransferases. Some restriction enzymes are sensitive to this, which will be indicated in the manufacturer’s product information. For these enzymes, grow the plasmid in commercially available E. coli strains in which Dam/Dcm methylation has been inactivated.
Plasmid storage
Keeping glycerol stocks of plasmid-containing bacteria is an ideal means of storage. Bacteria can be stored in media containing 15% glycerol at –70°C indefinitely [1]. Glycerol stocks preserve plasmid DNA and allow for quick production of additional plasmid. Fresh plasmid is produced by scraping the frozen glycerol stock with a sterile inoculating loop and streaking a fresh agar plate containing the appropriate selection antibiotic. The plate is incubated overnight at 37°C, and a single colony is selected and grown for purification. You can find additional protocols for making glycerol stocks in Green and Sambrook [1].
It is best to store purified DNA in nuclease-free buffer under slightly basic conditions, and in the presence of EDTA. EDTA inhibits nucleases by chelating divalent cations such as Mg2+ that are necessary for nuclease activity. IDT offers nuclease-free IDTE Buffers with 10 mM Tris, pH 7.5 or 8.0, 0.1 mM EDTA that are ideal for resuspension and storage of purified DNA. Plasmids can be stored in aliquots at –20°C, in a freezer that is not frost-free, for up to one year. Repeated freeze-thaw cycles will degrade nucleic acids and should be avoided. For storage longer than one year, storage at –70°C is recommended [2].
For additional information about cloning methods, download the IDT DNA Cloning Guide.
