
What is RNA sequencing?
RNA sequencing, or RNA-seq, is a powerful high-throughput, next generation sequencing (NGS) technique used to examine RNA in a single cell or a population of cells [1,2]. RNA-seq provides researchers with insights into gene expression levels, which allows them to draw conclusions about how a cell or group of cells functions. Using RNA-seq, researchers can determine if genes are being expressed at differing levels as well as if RNA is being transcribed abnormally (i.e., alternative splicing)[3].
How is RNA sequencing done?
An NGS RNA-seq workflow consists of four steps: nucleic acid extraction, enrichment, library preparation, and sequencing (Figure 1).
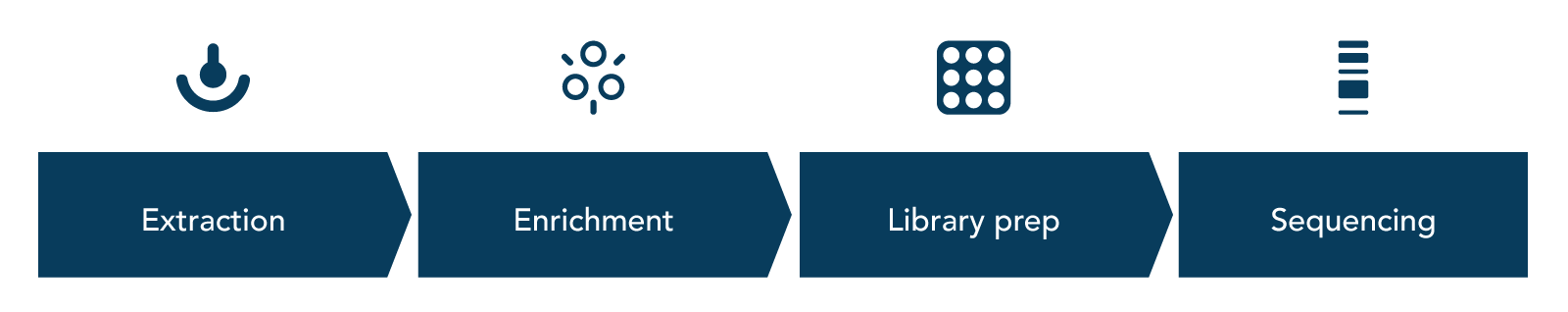
RNA extraction—RNA is released from cells. While there are various methods in which this can be done, to avoid the loss of precious RNA molecules, it is recommended that this step and those following are completed in an environment where RNases have been removed.
Enrichment/depletion—since the extraction step results in total RNA, in most cases, ribosomal RNA (rRNA) should be removed. rRNA can be up to 80% of RNA molecules present in a sample [4], meaning that when sequencing is completed, rRNA reads could become the majority of reads obtained—preventing researchers from gathering important information about other transcripts. There are two approaches that can be used to complete this step:
- mRNA enrichment—commonly used to enrich poly(A)-tailed mRNA by using olio(dT) beads that bind to the mRNA poly(A) tails. Once the beads are bound, what is left in the solution are the unwanted rRNA molecules. Once this rRNA rich solution is removed, the mRNA can be eluted from the beads. This approach is only recommended for samples in which mRNA molecules will have poly(A)-tails (i.e., eukaryotic samples).
- RNA depletion—the recommended approach for RNA removal if your sample does not have poly(A)-tailed mRNA molecules or if the mRNA molecules are degraded. While rRNA is a common target for RNA depletion other unwanted RNA molecules can also be targeted with this method—e.g., globin mRNA [5]. This approach also uses beads; however, RNA depletion beads are designed to bind the unwanted RNA molecules, leaving behind the desired mRNA.
Library preparation—if the starting RNA molecules are not too degraded and short read sequencing is going to be performed, then RNA molecules need to be fragmented. Fragmentation is followed by a reverse transcription reaction which converts the fragmented RNA into cDNA. Once this is completed, adapters and sample-specific indexes are added to the cDNA. The now indexed cDNA undergoes purification/clean-up, generating ready-to-sequence library molecules. Depending on the RNA-seq protocol that is followed, libraries can be stranded or non-stranded.
As the name implies, non-stranded RNA-seq protocols do not preserve information about the strand in which the mRNA transcript originated during cDNA synthesis. Without this information, downstream analyses of gene expression can be complicated, especially when the goal is to determine the expression levels of overlapping genes [6].
It is important that you consider the aim of your RNA-seq project when selecting a library prep kit, as some kits do provide chemistry that generates stranded RNA-seq libraries. To read more about stranded RNA library prep kits, visit the xGen RNA Library Prep Kit or xGen Broad-Range RNA Library Prep Kit webpage.
Sequencing—RNA-seq libraries can be prepared for single-end (SE) or paired-end (PE) sequencing. SE libraries provide reads from a single direction while PE libraries generate reads from both directions of a library molecule. PE libraries allow reads to be computationally merged post-sequencing, providing longer read fragments. The coverage or depth of sequencing depends on the objective of the RNA-seq project, but it is important to keep in mind that a sufficiently deep amount of coverage will be needed to perform a reliable analysis. You can read more about coverage and depth for NGS by downloading the next generation sequencing guide.
Types of RNA-seq
It is worth noting that there are several types of RNA-seq, each with their own unique workflow/objectives.
Examples of other types of RNA-seq include:
- Targeted RNA-seq focuses on expression levels for selected targets; for example, the exome, by incorporating a hybridization capture panel into the sample prep workflow. This approach provides deep sequencing of targeted regions while omitting the undesired regions that often result in a high number of reads not relevant to the research study.
- Single-cell RNA-seq (scRNA-seq) examines gene expression at the level of an individual cell rather than looking at the average gene expression across groups of cells. This technique allows researchers to look at gene expression heterogeneity within population of cells [2]. scRNA-seq workflows involve the isolation of single cells from a sample and a specialized RNA-preserving lysis step before RNA enrichment and cDNA synthesis [7,8].
- Small RNA-seq is a type of RNA-seq that isolates small noncoding RNAs (e.g., microRNAs). This approach allows researchers to investigate the role of small RNAs in post-transcriptional gene expression regulation, heterochromatin formation, and other cellular functions. In a small RNA-seq workflow, the targeted RNA molecules must undergo a size selection step to help ensure that the right species of RNAs are present in concentrations that are high enough for sequencing. This is done by gel electrophoresis, membrane purification, or bead selection [9].
Applications of RNA sequencing
RNA-seq can be performed on samples without having prior information about the genome, providing high levels of details about gene expression, alternative splicing, and other transcriptional features within a sample [3,10]. This makes it an industrious approach, and RNA-seq can be used for a number of applications including whole transcriptome research, novel transcript discovery, gene fusion research, and inherited disease and cancer research [11]. In the field of cancer research, RNA-seq has helped with the identification of cancer biomarkers through differential gene expression analyses [11] as well as the detection of gene fusions, which is another cancer-associated genetic marker [12].
To read more about how the application of RNA-seq in cancer research, download the application note RNA-seq for biomarker identification using the xGen Broad-Range RNA Library Prep Kit and xGen Custom Hyb Panels.
